
特點分析 | 中國創(chuàng)新醫(yī)藥研發(fā)企業(yè)赴美上市研究
專欄:行業(yè)新聞
發(fā)布日期:2018-02-09
閱讀量:7424
作者:
2017年9月22日,總部位于上海的再鼎醫(yī)藥(股票代碼:ZLAB)宣布完成首次公開發(fā)行美國存托股份并在美國納斯達克全球市場上市,該次發(fā)行募資總額約1.725億美元。當(dāng)日開盤價報24.25美元,首日漲幅超過55%,截至目前再鼎醫(yī)藥每股價格為27.11美元,總市值已達到13.37億美元[1]。而于2016年2月和7月在美國納斯達克市場上市的百濟神州和和黃醫(yī)藥[2]目前每股價格分別為94.50美元和35.04美元,總市值已分別達到42.97億和42.57億美元[3]。
與近幾年紛紛進行私有化退市的中概醫(yī)藥股不同的是,以再鼎醫(yī)藥、百濟神州等為代表的一批擁有獨特技術(shù)和產(chǎn)品的新銳創(chuàng)新藥研發(fā)公司逆勢在美國成功IPO并受到美國資本市場的青睞和追捧,這說明具有高潛力藥品管線(如腫瘤藥物)、強大研發(fā)能力和中國市場落地能力的中國創(chuàng)新藥物研發(fā)企業(yè)(“創(chuàng)新藥企”)正處于資本市場的下一個風(fēng)口。
一、赴美上市的中國創(chuàng)新藥企的主要特點
(一) 大多處于資金投入、未盈利階段,只能在美股上市
創(chuàng)新藥物研發(fā)周期長、風(fēng)險高,且投資巨大,所需要的投資動輒幾千萬或上億美元。對于一家無任何上市產(chǎn)品、絕大多數(shù)產(chǎn)品尚處于研發(fā)、臨床試驗階段的公司,很難做到盈利。隨著公司在研產(chǎn)品臨床研究的推進,對資金的需求會迅速加大,VC、PE的資金投入雖然對創(chuàng)新醫(yī)藥企業(yè)的發(fā)展起到了雪中送炭的作用,但經(jīng)歷了若干輪融資后的大多數(shù)中小型創(chuàng)新藥企仍無法確保公司有充足的資金將現(xiàn)有處于研發(fā)或臨床階段產(chǎn)品推向市場,同時還要面臨VC、PE盈利退出需求的壓力。在這種情況下,創(chuàng)新藥企無疑要從資本市場獲取更多的資金支持,以確保臨床研究和候選藥品上市申請的順利進行。但國內(nèi)A股的盈利門檻要求,讓仍在燒錢階段的創(chuàng)新藥企望而卻步。而美國資本市場對上市公司是否盈利、歷史沿革有沒有問題、經(jīng)營規(guī)模和資產(chǎn)有多大都不是特別在乎,它們關(guān)心的是企業(yè)未來的增長前景,是上市公司技術(shù)、產(chǎn)品、服務(wù)有沒有很大的潛在市場需求。美國資本市場對醫(yī)藥企業(yè)沒有盈利要求,國外投資者對無產(chǎn)品、專注研發(fā)并需要大量資金投入的初創(chuàng)生物制藥研發(fā)企業(yè)接受度更高,同其他境內(nèi)外資本市場相比較,在美國上市更容易獲得融資和取得良好的估值,這些是創(chuàng)新藥企多選擇在美國上市的主要原因。
(二) 大多擁有極具潛力價值的在研產(chǎn)品管線及強大的研發(fā)能力和團隊
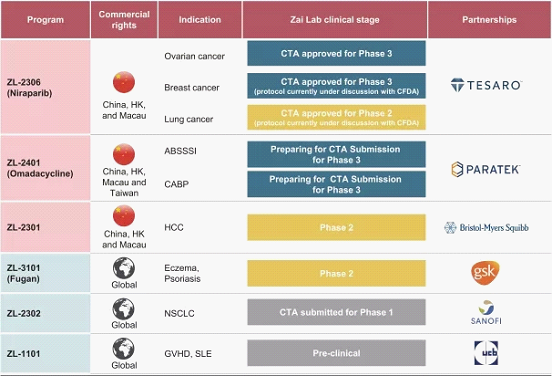
創(chuàng)新藥的研發(fā)要經(jīng)歷化合物篩選、臨床前試驗、臨床試驗、注冊申報等過程,其研發(fā)難度極其巨大,5000-10000個候選化合物之中才能有一個藥物最終上市,一個創(chuàng)新藥研發(fā)周期耗時長達10年,平均每個創(chuàng)新藥的研發(fā)費用達到數(shù)十億美元[4]。對于這些未盈利且需要巨大資金投入的創(chuàng)新藥研發(fā)企業(yè),要想獲得資本市場的認可和高估值,必須要有豐富的產(chǎn)品管線布局和強大的研發(fā)實力作為支撐。創(chuàng)新藥研發(fā)企業(yè)一般都是致力于研發(fā)具有創(chuàng)新性的、同類最佳的靶向類及最具獨特技術(shù)和資本市場熱點特征的藥物(比如免疫抗腫瘤藥物)。這些創(chuàng)新藥企雖然當(dāng)前還沒有上市產(chǎn)品,但研發(fā)管線大多非常強大,擁有數(shù)個臨床階段的在研藥物和數(shù)個臨床前產(chǎn)品。以再鼎醫(yī)藥為例,根據(jù)再鼎醫(yī)藥招股說明書[5]顯示,目前再鼎醫(yī)藥的在研產(chǎn)品管線從發(fā)現(xiàn)階段到臨床試驗后期階段共有6個產(chǎn)品布局,其中再鼎醫(yī)藥通過與TESARO達成戰(zhàn)略合作協(xié)議、獲得在中國市場的獨家研發(fā)和銷售權(quán)的藥品Niraparib(PARP抑制劑)已于2017年3月被FDA批準(zhǔn)在美上市,同時Niraparib(用于卵巢癌和乳腺癌治療)再鼎醫(yī)藥在中國市場的研發(fā)階段已經(jīng)進入臨床三期階段。在創(chuàng)新藥估值過程中,創(chuàng)新藥企的估值和在研管線的產(chǎn)品數(shù)量和所處階段基本上成正比,而如果一家創(chuàng)新藥企有1-2個在研產(chǎn)品獲得了FDA的上市批準(zhǔn)或已經(jīng)進入了臨床試驗后期階段并有很大可能性獲批上市,則其估值或市值將會獲得爆發(fā)性增長[6]。一家能夠獲得投資人、資本市場認可并給予高估值的創(chuàng)新藥企的產(chǎn)品管線必須豐富且分布合理,處于發(fā)現(xiàn)階段、臨床早期的產(chǎn)品能夠證明該創(chuàng)新藥企自身的研發(fā)能力、潛在的巨大價值,代表著未來技術(shù)市場的方向;而處于臨床試驗后期甚至已經(jīng)拿到上市批文的產(chǎn)品則證明了該創(chuàng)新藥企的研發(fā)實力、成功率,能夠讓投資人和資本市場看到了投資盈利和變現(xiàn)的前景。
創(chuàng)新藥企另一核心競爭力即是其擁有頂尖的行業(yè)領(lǐng)軍人物和強大的研發(fā)團隊實力。在美國上市的中國創(chuàng)新藥企的創(chuàng)始人或核心團隊,或是從海外歸來的頂尖科學(xué)家,或是出身于跨國藥企的資深高管。過去10年,國內(nèi)以創(chuàng)新為目的的人才引進計劃,推動了海外科學(xué)家的回國潮,也帶動了醫(yī)藥行業(yè)的密集而蓬勃的創(chuàng)業(yè)勢頭。這些具有海外歸國背景的創(chuàng)業(yè)團隊,不僅了解最新的科學(xué)技術(shù),也有國際化的人脈關(guān)系,在創(chuàng)業(yè)做創(chuàng)新藥的時候也更容易得到資本青睞。下圖是一些創(chuàng)新藥企的主要創(chuàng)始人、核心管理人員簡單背景介紹,足見創(chuàng)新藥企管理團隊的精英化構(gòu)成。
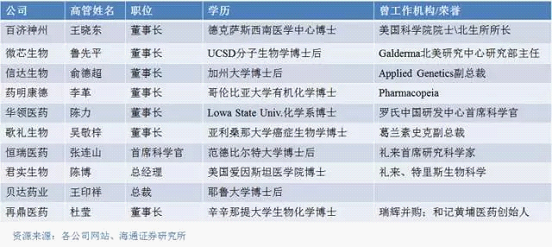
(三) 多數(shù)采用授權(quán)許可為主,自主研發(fā)為輔的商業(yè)模式
依照中國食品藥品監(jiān)督管理總局(CFDA)2016年2月頒布的《化學(xué)藥品注冊分類改革工作方案》,創(chuàng)新藥是指境內(nèi)外均未上市的創(chuàng)新藥,指含有新的結(jié)構(gòu)明確的、具有藥理作用的化合物,且具有臨床價值的藥品。新藥研發(fā)是一個逐漸積累的過程,即使在兼具科研機構(gòu)、資金、頂尖醫(yī)院等各種豐富資源的西方國家,一個新藥從最初發(fā)現(xiàn)到最終上市也要經(jīng)歷數(shù)十年漫長的時間積累和無數(shù)的失敗。擁有一個具有完全自主知識產(chǎn)權(quán)的創(chuàng)新藥固然重要,但也要考慮創(chuàng)新藥自身特有的難度和規(guī)律。 “盡管first-in-class聽上去令人激動,但實際上風(fēng)險太高,很難為投資人提供穩(wěn)健的回報。在這種情況下,我們首先關(guān)注的是新藥的安全性,其次追求有效性在同類藥里最優(yōu)(best-in-class)。”徐諾藥業(yè)董事長徐英霖說。來自于Nature Reviews Drug Discovery的報告《Trends in clinical success rates》指出,合作項目的成功率是遠遠高于獨立項目的。因此,即便是跨國制藥巨頭也是將合作開發(fā)作為風(fēng)險管控的一種有效方式。目前創(chuàng)新藥企一個基本的模式是,通過與全球頂尖跨國公司或研發(fā)機構(gòu)建立戰(zhàn)略合作關(guān)系,從而在全球范圍內(nèi)甄選較為成熟的臨床候選藥物,共同進行臨床研究,通過內(nèi)部研發(fā)和授權(quán)許可并行的方式,建立全面的產(chǎn)品線。而再鼎醫(yī)藥正是這一模式的開創(chuàng)者和踐行者,根據(jù)再鼎醫(yī)藥招股說明書顯示,目前再鼎醫(yī)藥的在研產(chǎn)品管線6個產(chǎn)品中,其中來自GSK、賽諾菲以及UCB的三個產(chǎn)品再鼎均取得的是相關(guān)藥品在全球的獨家臨床開發(fā)、注冊、生產(chǎn)以及銷售權(quán),而與TESARO、PARATEK、BMS等合作的三個產(chǎn)品則取得的是大中華區(qū)權(quán)益,并且多項產(chǎn)品已經(jīng)進入了臨床研發(fā)的中后期。下圖為再鼎醫(yī)藥的產(chǎn)品研發(fā)管線:
二、赴美上市的中國創(chuàng)新藥企遇到的主要法律問題及風(fēng)險
(一)紅籌架構(gòu)和境內(nèi)身份股東出境問題
在美國上市的中概股公司采用的紅籌架構(gòu)主要為VIE架構(gòu)和股權(quán)控制架構(gòu)。赴美上市的中國創(chuàng)新藥企多采用股權(quán)控制架構(gòu),主要是因為創(chuàng)新藥的研發(fā)屬于外商投資鼓勵類行業(yè),無需采用VIE架構(gòu)間接控制,同時,創(chuàng)新藥企的創(chuàng)始人多數(shù)為具有外籍的海外歸國人員以及主要投資人大多為境外基金。不過,由于外商投資企業(yè)的業(yè)務(wù)經(jīng)營需要受制于《外商產(chǎn)業(yè)指導(dǎo)目錄》關(guān)于外商投資領(lǐng)域的限制,雖然股權(quán)控制架構(gòu)無VIE架構(gòu)固有的法律和道德風(fēng)險,但股權(quán)控制架構(gòu)的外資創(chuàng)新藥企將不能從事一些特定領(lǐng)域的醫(yī)藥或技術(shù)的研究,如涉及人體干細胞、基因技術(shù)的醫(yī)藥及技術(shù)研發(fā)業(yè)務(wù)[7]。
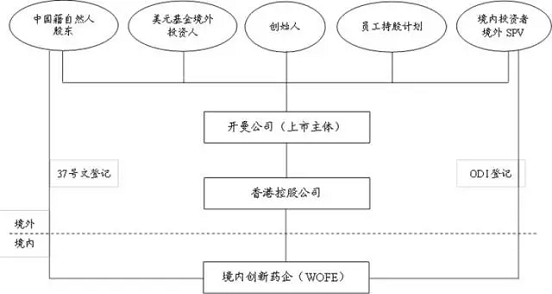
此外,雖然赴美上市創(chuàng)新藥企的股東大多為外籍人士或境外美元基金,但也會有一些公司的股東為中國籍人士或在公司融資階段引入了一些人民幣基金或境內(nèi)投資者,在美國上市過程中,需要解決這些境內(nèi)身份股東的持股出境問題或者說紅籌化重組問題。中國自然人股東持股權(quán)益出境通常是通過辦理外管37號文[8]登記,在目前的政策環(huán)境下辦理渠道還是比較便捷和暢通。同時,創(chuàng)新藥企需要辦理37號文登記的中國籍自然人股東通常持股比例不高,即便是辦理外管37號文有一定難度,也可通過委托其他外籍創(chuàng)始人或者已經(jīng)完成外管37號登記的中國籍自然人股東進行代持解決。但對于境內(nèi)機構(gòu)投資人,由于機構(gòu)投資人在創(chuàng)新藥企的數(shù)輪融資中投入的都是真金白銀且持股比例通常也較高,在紅籌重組中很難用代持方式解決出境問題。境內(nèi)機構(gòu)投資人的境外持股方式通常為履行中國法律對外投資法律規(guī)定下的境內(nèi)機構(gòu)境外投資(“ODI”)監(jiān)管流程,即辦理當(dāng)?shù)匕l(fā)改委、商務(wù)部門和外匯管理局的ODI備案登記,通過在境外設(shè)立自己的持股平臺(SPV)或其在境外已有的投資實體持有創(chuàng)新藥企境外開曼控股公司(將來的上市主體)所發(fā)行的股份,并將自己在境內(nèi)創(chuàng)新藥企的持股轉(zhuǎn)讓給境外控股公司[9]。包含境內(nèi)持股結(jié)構(gòu)的創(chuàng)新藥企紅籌化重組架構(gòu)如下圖:
但是,從2016年開始,隨著中國對境內(nèi)機構(gòu)境外投資的監(jiān)管政策逐步收緊,特別是在2017年8月國辦發(fā)〔2017〕74號[10]發(fā)布之后,“在境外設(shè)立無具體實業(yè)項目的股權(quán)投資基金或者投資平臺”被列為限制開展的境外投資并需要境外投資主管部門核準(zhǔn),境內(nèi)機構(gòu)通過ODI在境外設(shè)立持股平臺SPV方式實現(xiàn)權(quán)益出境將面臨著更嚴的監(jiān)管態(tài)勢和不確定性。國辦發(fā)〔2017〕74號文的前述規(guī)定影響的不僅僅是具有境內(nèi)投資主體的創(chuàng)新藥企的紅籌化重組,而是將影響到所有通過設(shè)立境外SPV方式從事海外投資和并購的投資機構(gòu),雖然如何定義“無具體實業(yè)項目的股權(quán)投資基金或者投資平臺”、如何限制及審核尺度、期限都還比較模糊,有待于這一文件的具體執(zhí)行細則或出臺相關(guān)法規(guī)予以落實,但可以預(yù)見在未來一個時期,以境外上市為目的的紅籌化重組將面臨更多的受政策法規(guī)不確定因素影響的風(fēng)險。
(二)中國創(chuàng)新醫(yī)藥行業(yè)監(jiān)管政策、法規(guī)的影響
醫(yī)藥行業(yè),尤其是創(chuàng)新藥行業(yè),是一個受政策和監(jiān)管法規(guī)影響比較大的行業(yè),藥品監(jiān)督管理政策將直接影響創(chuàng)新藥企的創(chuàng)新動力和運營發(fā)展。中國創(chuàng)新藥行業(yè)整體水平不高一方面是由于創(chuàng)新藥企自身科研水平和資金不足,風(fēng)險抵抗力較弱; 另一方面,中國長期以來缺乏激勵創(chuàng)新的機制和配套政策以及落后的監(jiān)管政策法規(guī)嚴重制約了創(chuàng)新藥企的發(fā)展和新藥研發(fā)動力。因此,為了推動我國藥品特別是重大新藥研發(fā),促進醫(yī)療器械產(chǎn)業(yè)升級發(fā)展,從2015年8月國務(wù)院公布《關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》以來,中國陸續(xù)出臺了一系列的深化醫(yī)藥評審制度改革和鼓勵藥品創(chuàng)新的政策和法規(guī),特別是2017年10月國務(wù)院《關(guān)于深化審評審批制度改革鼓勵藥品醫(yī)療器械創(chuàng)新的意見》(“《意見》”)的正式公布,將對創(chuàng)新醫(yī)藥行業(yè)的快速發(fā)展產(chǎn)生深遠的影響和巨大的促進作用。
《意見》中惠及創(chuàng)新藥企的主要改革政策和措施主要體現(xiàn)在:(1)改革臨床試驗管理,臨床試驗機構(gòu)從原來的資格認定制改為備案制;改革臨床試驗的審批,由過去的明示許可改為默示許可;(2)優(yōu)化審評審批,明確附帶條件批準(zhǔn)的情形,罕見病用藥和臨床急需藥,可以附帶條件批準(zhǔn);此外,藥品與原料、輔料、包裝材料今后不單獨審批,而是跟制劑關(guān)聯(lián)審評審批;(3)加強創(chuàng)新權(quán)益保護,探索建立藥品專利鏈接制度,開展藥品專利期的補償試點,完善和落實藥品數(shù)據(jù)保護以及建立上市藥品目錄集;(4)藥品監(jiān)管一定要突出上市許可持有人制度,明確上市許可持有人的責(zé)任。[11]
《意見》對創(chuàng)新藥企的研發(fā)和經(jīng)營將帶來新的機遇。例如在臨床試驗的數(shù)據(jù)使用領(lǐng)域,以前中國法規(guī)要求在我國上市的藥品必須要在我國開展臨床試驗,而此次《意見》中提出了在境外多中心臨床機構(gòu)取得的臨床試驗數(shù)據(jù),符合我國藥品注冊要求的,可以用于在我國申報注冊。赴美上市的創(chuàng)新藥企大多都在美國、澳大利亞、歐盟等境外醫(yī)藥研究發(fā)達市場開展藥品注冊和臨床試驗,這樣就可以減少重復(fù)臨床試驗,提高效率,降低研發(fā)費用,大大縮短新藥申請注冊和最終上市的時間。在享受藥政改革紅利的同時,創(chuàng)新藥企也將面臨更多的挑戰(zhàn)或責(zé)任。比如,《意見》再次強調(diào)了臨床試驗數(shù)據(jù)的真實性要求,堅持嚴肅查處數(shù)據(jù)造假行為,規(guī)定臨床試驗委托協(xié)議簽署人和臨床試驗研究者是臨床試驗數(shù)據(jù)的第一責(zé)任人,需要對臨床試驗數(shù)據(jù)的可靠性承擔(dān)法律責(zé)任。如果存在真實性問題的,將依法追究相關(guān)非臨床研究機構(gòu)和臨床試驗機構(gòu)責(zé)任人、虛假報告提供責(zé)任人、注冊申請人及合同研究組織責(zé)任人的責(zé)任。
需要指出的是,《意見》及多數(shù)藥政改革文件都是一些綱領(lǐng)性指導(dǎo)文件,尚未形成正式的法律法規(guī),目前的《藥品管理法》和《藥品注冊管理辦法》等適用于創(chuàng)新藥企的法律法規(guī)還大多都處于修訂、征求意見稿或試點階段,因此將來正式頒布修訂的《藥品管理法》和《藥品注冊管理辦法》對上述藥政改革文件的落實和執(zhí)行情況還存在一定的不確定性。
(三)創(chuàng)新藥企知識產(chǎn)權(quán)的保護及相關(guān)法律問題
醫(yī)藥產(chǎn)業(yè)對專利保護的依賴程度遠遠高于其他技術(shù)領(lǐng)域。美國著名經(jīng)濟學(xué)家曼斯菲爾德曾指出,“如果沒有專利保護,60%的新藥不會被發(fā)明出來”。這是因為新藥研發(fā)具備“高投入、高風(fēng)險、長周期”的特點,前期需要進行大量的臨床試驗,后續(xù)又需經(jīng)歷復(fù)雜和漫長的藥品注冊程序,而相對于原研藥而言,仿制藥的研發(fā)周期相對較短,審批程序相對較為容易,使其能夠以更低廉的價格進入市場。因此,如果沒有健全的專利制度保證原研藥企業(yè)研發(fā)投入的回報率,原研藥企業(yè)的創(chuàng)新動力將大受影響,不利于醫(yī)藥產(chǎn)業(yè)的持續(xù)創(chuàng)新。
根據(jù)我國《專利法》的規(guī)定,發(fā)明專利權(quán)的保護期為二十年,自專利申請之日起計算。在醫(yī)藥領(lǐng)域,藥品發(fā)明專利的保護對象主要包括新化合物、新制備方法、新藥物制劑、新用途等。其中,化合物專利保護的是活性藥物成分(Active Pharmaceutical Ingredient, API),是最能體現(xiàn)藥物核心競爭力的專利。如上所述,化學(xué)新藥的誕生要經(jīng)歷“篩選”和“開發(fā)”兩個階段。在篩選階段完成后,接下來就是漫長的臨床試驗階段了。因此,有經(jīng)驗的藥企一般會隨著研發(fā)的進程分階段申請先導(dǎo)化合物、通式化合物、優(yōu)選化合物、制劑、晶型、制備方法、新用途、組合物專利等,以便最大程度地利用專利保護帶來的優(yōu)勢。即使如此,藥品專利的審查通常需要比一般專利授權(quán)審查更長的時間,再加之漫長的臨床試驗和審評審批程序,很多創(chuàng)新藥在上市時,專利保護期已所剩無幾。此外,《藥品注冊管理辦法》規(guī)定,仿制藥審批可以在專利期結(jié)束前兩年申請。因此,專利保護期屆滿后,仿制藥能夠以相對低廉的價格迅速進入市場。
為了促進本國醫(yī)藥行業(yè)的創(chuàng)新發(fā)展,許多國家都專門針對創(chuàng)新藥設(shè)置了相關(guān)專利期延長制度,用以彌補專利生效到新藥獲批之間的時間損失。我國現(xiàn)行的《專利法》中尚未規(guī)定專利期延長制度,但在十月份頒布的《意見》中,國務(wù)院已提出選擇部分新藥開展藥品專利期限補償制度試點,對因臨床試驗和審評審批延誤上市的時間,給予適當(dāng)專利期限補償。我國《專利法》正在進行第四次修改,該藥品專利期限補償?shù)木唧w規(guī)定如何落地尚不明確,創(chuàng)新藥企業(yè)應(yīng)密切關(guān)注相關(guān)法律動態(tài),并及時調(diào)整研發(fā)戰(zhàn)略。
在醫(yī)藥領(lǐng)域,專利鏈接機制也是一項非常值得一提的知識產(chǎn)權(quán)制度。專利鏈接是指將仿制藥注冊與原研藥專利保護期“鏈接”,即仿制藥注冊申請應(yīng)當(dāng)考慮先前已上市參比制劑的專利情況。健全的專利鏈接制度對于促進藥品創(chuàng)新和平衡仿制藥發(fā)展具有重要作用。我國通過現(xiàn)行的《藥品注冊管理辦法》建立了初步的專利鏈接制度,但由于藥品專利信息檢索系統(tǒng)尚未健全、CFDA和國家知識產(chǎn)權(quán)局(SIPO)之間未形成有效的溝通機制以及CFDA并不注重專利侵權(quán)審查等問題,其尚未發(fā)揮應(yīng)有的作用。
2017年5月,CFDA發(fā)布了《關(guān)于鼓勵藥品醫(yī)療器械創(chuàng)新保護創(chuàng)新者權(quán)益的相關(guān)政策(征求意見稿)》(“《征求意見稿》”),致力于進一步在我國建立專利鏈接制度。《征求意見稿》要求,相關(guān)藥品專利權(quán)人認為申請人侵犯其專利權(quán)的,應(yīng)在接到申請人的不侵權(quán)聲明后20天內(nèi)向司法機關(guān)提起專利侵權(quán)訴訟。創(chuàng)新藥企為了推遲仿制藥上市時間,似乎必須在20天內(nèi)提起專利侵權(quán)訴訟,而專利侵權(quán)訴訟又通常會導(dǎo)致并行的專利無效宣告程序,甚至發(fā)展至專利無效行政訴訟。可見,專利鏈接制度的引入可能對創(chuàng)新藥企的專利穩(wěn)定性產(chǎn)生影響,而且訴訟等重大情況也會影響企業(yè)的IPO戰(zhàn)略。《意見》也提出要探索建立專利鏈接制度,同時規(guī)定了對挑戰(zhàn)專利成功藥品注冊申請人提交的自行取得且未披露的試驗數(shù)據(jù)和其他數(shù)據(jù),給予一定的數(shù)據(jù)保護期,這一規(guī)定一方面可以激勵仿制藥企業(yè)進行專利挑戰(zhàn),另一方面也會增加原研藥企業(yè)專利狀態(tài)的不穩(wěn)定性。
(四)知識產(chǎn)權(quán)許可的法律險
在自主研發(fā)和擁有專利以外,創(chuàng)新藥企也會采取license-in(授權(quán)引進)方式獲得實施相關(guān)專利和專有技術(shù)的許可,用于研發(fā)、生產(chǎn)和商業(yè)化原研藥物。例如,根據(jù)百濟神州招股書披露,百濟神州的一款名為BGB-A317候選藥物相關(guān)的三項美國專利的專利權(quán)人Ono Pharmaceutical Co和被許可方Bristol-Myers Squibb Co,正在與Merck & Co.就該三項美國專利進行侵權(quán)訴訟,被告在侵權(quán)訴訟中挑戰(zhàn)了該三項美國專利的有效性。如果該三項美國專利的有效性被予以維持,而百濟神州計劃在美國生產(chǎn)、銷售該藥物,則其需向Bristol-Myers Squibb Co取得美國境內(nèi)的分許可,或者如果百濟神州計劃在其他國家生產(chǎn)、銷售該藥物,則其需就三項專利在該國家的同族專利獲得許可。
知識產(chǎn)權(quán)許可可能直接來自于藥物專利權(quán)人,也可能來自于具有分許可權(quán)限的藥物專利被許可方。而來自知識產(chǎn)權(quán)許可合同的法律風(fēng)險,可能嚴重制約創(chuàng)新藥企的研發(fā)進程,甚至?xí)箘?chuàng)新藥企喪失臨床候選藥物的研發(fā)能力或法律權(quán)利。例如,如果專利權(quán)人為比較強勢的跨國醫(yī)藥企業(yè),有時在簽署許可協(xié)議前,被許可方很難推動對專利權(quán)人相關(guān)專利的盡職調(diào)查,尤其是專有技術(shù)的盡職調(diào)查。而跨國企業(yè)內(nèi)部關(guān)聯(lián)公司之間的專利權(quán)屬關(guān)系復(fù)雜,有可能專利權(quán)人是一家子公司,而許可協(xié)議簽署方卻是另一實體,如果許可協(xié)議中事先沒有約定許可方授予許可的權(quán)利,就可能導(dǎo)致爭議,甚至終止許可合同。另外,在實施專利的過程中很可能需要利用相關(guān)專有技術(shù),如果許可協(xié)議中沒有清晰約定專有技術(shù)許可或者沒有詳細列舉專有技術(shù)清單,也可能產(chǎn)生商業(yè)秘密侵權(quán)爭議等隱患。
此外,如果創(chuàng)新藥企在談判時拿到的僅是大中華區(qū)權(quán)益,該候選藥物在全球的開發(fā)進度和成功與否會決定其在中國市場的命運。如果該授權(quán)項目在國外由于資金、合作伙伴、內(nèi)部規(guī)劃等原因被終止,或者沒有通過FDA或EMEA的批準(zhǔn),大中華區(qū)的項目也很可能會因此而受到影響。
(五)創(chuàng)新藥企對第三方的依賴及相關(guān)法律問題
目前赴美上市的創(chuàng)新藥企多數(shù)為“小而美”的精英化團隊運營的中小型創(chuàng)新性企業(yè),他們能夠發(fā)揮小型企業(yè)的優(yōu)勢,充分與外部第三方合作,利用合同研究組織(CRO),受托生產(chǎn)企業(yè)(CMO)以及和跨國藥企、研究機構(gòu)的合作,充分借用自身的技術(shù)優(yōu)勢,提升公司的研發(fā)效率。不過,該種運營模式對于小型創(chuàng)新藥企來說也是把“雙刃劍”,在充分利用合作方的資金、產(chǎn)品、技術(shù)、市場等優(yōu)勢提升自身研發(fā)實力的同時,合作方及合作產(chǎn)品在法律方面的一些重大瑕疵、缺陷也可能會給創(chuàng)新藥企帶來巨大的法律風(fēng)險。
2016年5月26日,國務(wù)院出臺了《藥品上市許可持有人制度試點方案》,開始在10個省市開展上市許可持有人制度(MAH)試點,突破了以往的上市許可與生產(chǎn)許可的“捆綁制”。此次《意見》提出推動上市許可持有人制度全面實施,有利于總結(jié)試點經(jīng)驗,從制度上保證創(chuàng)新藥企能以上市許可持有人的身份享有技術(shù)創(chuàng)新所帶來的最終市場收益,極大的促進創(chuàng)新藥企的研發(fā)動力。
在上市許可持有人制度下,如果作為上市許可持有人的創(chuàng)新藥企不具備相應(yīng)生產(chǎn)資質(zhì),則須委托具備資質(zhì)的藥品生產(chǎn)企業(yè)生產(chǎn)批準(zhǔn)上市的藥品。CMO應(yīng)當(dāng)為依法設(shè)立、持有相應(yīng)藥品生產(chǎn)范圍的《藥品生產(chǎn)許可證》以及藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP)認證證書的藥品生產(chǎn)企業(yè)。申請人、持有人是藥品上市許可的責(zé)任主體,承擔(dān)藥品全生命周期的安全性有效性保證義務(wù),包括注冊、生產(chǎn)、流通、監(jiān)測和評價、質(zhì)量追溯、信息公開等,雖然某些義務(wù)可以與生產(chǎn)企業(yè)進行約定,但最終責(zé)任應(yīng)該由申請持有人承擔(dān)。而如果受托生產(chǎn)企業(yè)未能遵守法律規(guī)定的生產(chǎn)設(shè)施檢查要求和質(zhì)量管理規(guī)范,或受到監(jiān)管部門的處罰,將會給委托方(創(chuàng)新藥企)帶來嚴重的法律風(fēng)險和法律責(zé)任,直接導(dǎo)致新藥研發(fā)和生產(chǎn)進程受阻。這將要求創(chuàng)新藥品持有人需要具備較強的風(fēng)險承擔(dān)和管控能力。
三、結(jié)語
創(chuàng)新醫(yī)藥研發(fā)產(chǎn)業(yè)是一個能夠持續(xù)增長,潛力巨大的金礦,在國家鼓勵創(chuàng)新的政策扶持下,已經(jīng)迎來發(fā)展黃金期。而赴美IPO、獲得國際資本市場的認可和資金支持,則會使中國創(chuàng)新藥企獲得更多加速研發(fā)的資源和力量,只要是在創(chuàng)新發(fā)展的過程中,充分重視有關(guān)法律風(fēng)險和規(guī)避潛在的重大法律問題,在醫(yī)藥改革的大背景下,中國創(chuàng)新醫(yī)藥研發(fā)產(chǎn)業(yè)必將步入發(fā)展的快車道。在祝賀再鼎醫(yī)藥、百濟神州這些新銳創(chuàng)新藥企成功登陸納斯達克并獲得巨大成功的同時,我們也希望看到越來越多的中國創(chuàng)新藥企借助境內(nèi)外資本市場的力量,加快藥品創(chuàng)新,讓更多新藥能夠問世,造福中國乃至全球的患者,并真正實現(xiàn)中國從醫(yī)藥大國向醫(yī)藥強國的突破。
[1] 參見http://quote.eastmoney.com/us/zlab.html?StockCode=zlab,最后訪問日期:2017年12月5日
[2] 現(xiàn)名為“和記中國醫(yī)療科技”
[3] 參見http://quote.eastmoney.com/us/BGNE.html 和http://quote.eastmoney.com/us/hcm.html?StockCode=hcm ,最后訪問日期:2017年12月5日
[4] 《創(chuàng)新藥價值評估初探--創(chuàng)新藥行業(yè)深度研究之二》-興證醫(yī)藥團隊
[5] 參見https://www.sec.gov/Archives/edgar/data/1704292/000119312517290496/d358845d424b4.htm
[6] 例如 CELATOR(CPXX.O)公司 2016年3月15日宣布其研制的納米新劑型抗癌藥CPX-351在高危急性髓細胞白血病三期臨床試驗上取得了積極成果。CPXX臨床試驗結(jié)果顯示,CPX-351與標(biāo)準(zhǔn)療法相比,死亡風(fēng)險降低31%,受此利好消息影響,股價單日暴漲432.1%。
文章鏈接:中國制藥網(wǎng) http://www.zyzhan.com/news/detail/dy55489_p2.html
[7] 依照2017年版《外商投資產(chǎn)業(yè)指導(dǎo)目錄》,“人體干細胞、基因診斷與治療技術(shù)開發(fā)和應(yīng)用”被列為《外商投資準(zhǔn)入特別管理措施(負面清單)》中的“禁止外商投資產(chǎn)業(yè)目錄”。
[8] 匯發(fā)[2014]37號 《國家外匯管理局關(guān)于境內(nèi)居民通過特殊目的公司境外投融資及返程投資外匯管理有關(guān)問題的通知》
[9] 如果創(chuàng)新藥企的境內(nèi)投資人是有著境內(nèi)境外平行基金結(jié)構(gòu)安排的人民幣基金,該人民幣基金的境內(nèi)權(quán)益出境可以由其境外美元基金在境外認購開曼控股公司的同比例的股份,而人民幣基金在境內(nèi)投資權(quán)益退出(但通常情況下,人民幣基金和境外美元基金的LP要相一致或具備關(guān)聯(lián)關(guān)系,操作起來也會比較復(fù)雜)。
[10] 《國務(wù)院辦公廳轉(zhuǎn)發(fā)國家發(fā)展改革委商務(wù)部人民銀行外交部關(guān)于進一步引導(dǎo)和規(guī)范境外投資方向指導(dǎo)意見的通知》,2017年8月18日發(fā)布。
[11] 根據(jù)《意見》和2017年10月9日上午10時國家食品藥品監(jiān)督管理總局(“CFDA”)藥品醫(yī)療器械審評審批改革鼓勵創(chuàng)新工作新聞發(fā)布會CFDA副局長吳湞就相關(guān)問題回答記者提問相關(guān)內(nèi)容進行整理。